The ERV component of Lentivirus-GLUE
Endogenous retroviruses (ERVs) are retrovirus-derived DNA sequences that occur in the germline genomes of host species. ERVs arise when retroviruses infect germline cells (i.e. spermatozoa, eggs, or early embryonic cells) so that integrated retrovirus DNA is vertically inherited as a novel host allele.
ERVs provide a unique source of retrospective information about the long-term history and retroviruses and their hosts. ERVs derived from lentiviruses are very rare, and for decades they were assumed not to occur. However, as described here, whole genome sequencing of vertebrates in the 2000s led to the discovery of ERVs derived from lentiviruses in a variety of species. These discoveries allowed the timeline of lentivirus evolution to be reliably calibrated.

Species containing lentivirus ERVs: left to right - leporids (family Leporidae); lemurs (family Lemuridae); Sunda flying lemur (Galeopterus variegatus); mink (Neovison vison)
Progress in characterising ERVs has been hampered by the challenges encountered attempting to analyse their fragmentary and degenerated sequences. This greatly complicates comparative genomic analysis. Lentivirus-GLUE aims to address these issues for lentiviral ERVs. it applies a set of programmatic principles for systematically organising and refining the ERV 'fossil record',
This page provides background information about the ERV component of Lentivirus-GLUE, and direct links to specific data items.
Paleovirus-specific schema extensions in Lentivirus-GLUE
The paleovirus component of Lentivirus-GLUE extends GLUE's core schema to allow the capture of ERV-specific data. These schema extensions are defined in this file and comprise two additional tables: 'locus_data' and 'refcon_data'. Both tables are linked to the main 'sequence' table via the 'sequenceID' field.
The 'locus_data' table contains information pertaining to individual ERV sequences: e.g. species in which they occur, genome assembly version, genomic location(i.e. scaffold, location coordinates, and orientation).
The 'refcon_data' table contains information pertaining to our ERV reference sequences, which we have constructed in an effort to reconstruct, as closely as possible, the sequences of the progenitor viruses that gave rise to ERVs.
Reference sequences and genome features
For each lentiviral paleovirus species, we have defined a 'master' reference sequence, as follows:
- Rabbit endogenous lentivirus K: (RELIK con)
- Prosimian immunodeficiency virus 1: (PSIV1-con)
- Prosimian immunodeficiency virus 2: (PSIV2-con)
- Mustelid endogenous lentivirus: (MELV)
- Dermopteran endogenous lentivirus: (DELV)
- Springhare endogenous lentivirus: (SpELV)
The paleovirus reference sequences represent the efforts of researchers to reconstruct, as closely as possible, the genomes of the ancestral lentiviruses that gave rise to ERV lineages. All reference sequences are linked to auxiliary data in tabular format.
We defined the known locations of lentivirus genome features on paleovirus reference sequences. (see here). Putative ancestral open reading frames (ORFs) encoding accessory genes have been identified in many endogenous lentiviruses. In some cases these represent clear homologs of the accessory genes found in exogenous lentiviruses, but in others it remains unclear whether they are homologs of previously described accessory genes or distinct genes. We therefore extended the set of lentivirus genome features defined in our core project to include these putatively distinct genes.
Lentivirus ERV sequences and sequence-associated data
The tabular file contains information about the genomic location of each ERV. ERVs were classified by comparison to a reference library of lentivirus polypeptide sequences library designed to represent the known diversity of lentiviruses - this includes extinct lineages represented only by endogenous viral elements (ERVs).
ERV sequences were recovered from whole genome sequence (WGS) assemblies via database-integrated genome screening (DIGS) performed in vertebrate genome assemblies downloaded from NCBI's genomes resource.
- Sequence-associated data in tabular format can be found here.
- Nucleotide level data in FASTA format (individual files) can be found here.
- The complete list of vertebrate genomes screened can be found here.
- The set of lentivirus polypeptide sequences used as probes can be found here.
- The final set of lentivirus and ERV polypeptide sequences used as references can be found here.
- Input parameters for screening using the DIGS tool can be found here.
Multiple sequence alignments (MSAs)
Multiple sequence alignments (MSAs) are the basic currency of comparative genomic analysis. MSAs constructed in this study are linked together using GLUE's constrained MSA tree data structure.
A 'constrained MSA' is an alignment in which the coordinate space is defined by a selected reference sequence. Where alignment members contain insertions relative to the reference sequence, the inserted sequences are recorded and stored (i.e. sequence data is never deleted).
GLUE projects have the option of using a data structure called an alignment tree to link constrained MSAs representing different taxonomic levels, and we've used this approach in Lentivirus-GLUE.
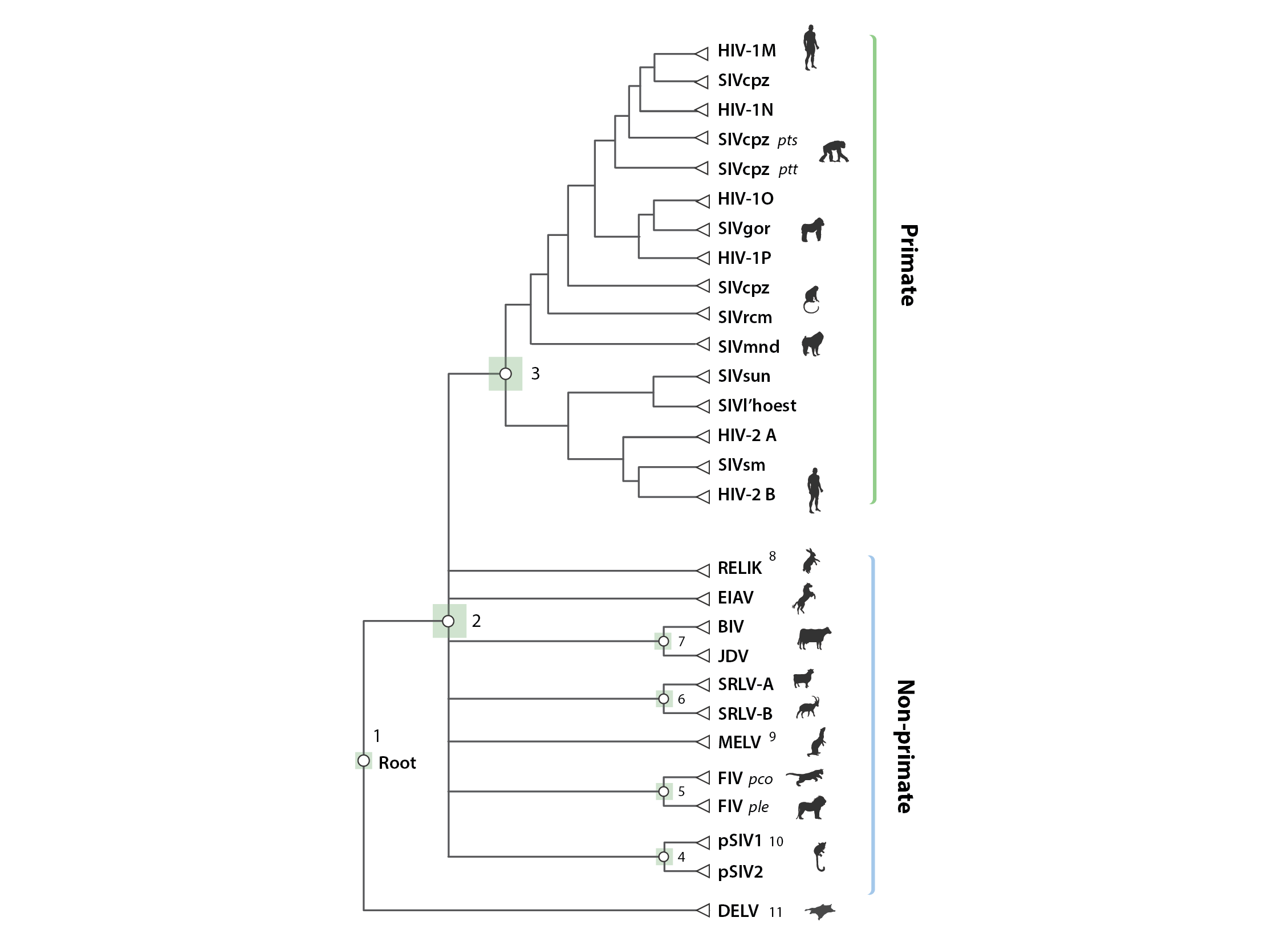
The schematic figure above shows the 'alignment tree' data structure currently implemented in Lentivirus-GLUE. We used an alignment tree data structure to link alignments, via a set of common reference sequences. The root alignment contains reference sequences for major clades, whereas all children of the root inherit at least one reference from their immediate parent. Thus, all alignments are linked to one another via our chosen set of master reference sequences.
Alignments in the project include:
- A single ‘root’ alignment constructed to represent proposed homologies between representative members of major lentivirus lineages (including extinct lineages represented only by ERVs).
- ‘Genus-level’ alignments constructed to represent proposed homologies between the genomes of representative members of specific lentivirus genera and ERV reference sequences.
- ‘Tip’ alignments in which all taxa are derived from a single ERV lineage.
ERV Nomenclature
We have applied a systematic approach to naming ERVs. Each element was assigned a unique identifier (ID) constructed from a defined set of components.

The first component is the classifier ‘ERV’ (endogenous retrovirus).
The second component is a composite of two distinct subcomponents separated by a period: (i) the name of ERV group; (ii) a numeric ID that uniquely identifies the insertion. The numeric ID is an integer identifies a unique insertion locus that arose as a consequence of an initial germline infection. Thus, orthologous copies in different species are given the same number.
The third component of the ID defines the set of host species in which the ortholog occurs.
Phylogenetic trees
We used GLUE to implement an automated process for deriving midpoint rooted, annotated trees from the alignments included in our project.
Trees were constructed at distinct taxonomic levels:
- Root phylogeny (Rep)
- ERV lineage-level phylogenies
Related Publications
Singer JB, Thomson EC, McLauchlan J, Hughes J, and RJ Gifford
(2018)
GLUE: A flexible software system for virus sequence data.
BMC Bioinformatics
[view]
Zhu H, Dennis T, Hughes J, and RJ Gifford
(2018)
Database-integrated genome screening (DIGS): exploring genomes heuristically using sequence similarity search tools and a relational database.
[preprint]
Gifford RJ, Blomberg B, Coffin JM, Fan H, Heidmann T, Mayer J, Stoye J, Tristem M, and WE Johnson
(2018)
Nomenclature for endogenous retrovirus (ERV) loci.
Retrovirology
[view]
Gifford RJ
(2012)
Viral evolution in deep time - Lentiviruses and mammals.
Trends in Genetics
[view]
Gifford RJ, Katzourakis A, Tristem M, Pybus, OG, Winters M, and RW. Shafer.
(2008)
A transitional endogenous lentivirus from the genome of a basal primate and implications for lentivirus evolution.
PNAS
[view]
Katzourakis A, Tristem M, Pybus OG, and RJ. Gifford (2007)
Discovery and analysis of the first endogenous lentivirus.
PNAS
[view]